Active Grants — Cancer and Cell Aging Platform
Title: The role of mutant KRAS isoforms in lung cancer
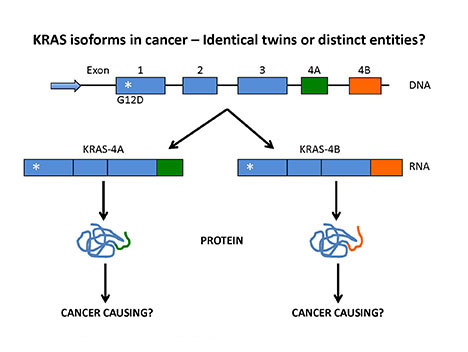
The KRAS gene encodes two distinct flavors depending on whether exon 4A or 4B is used in the RNA. The project investigates whether KRAS4A or KRAS4B are responsible for tumor initiation.
Investigative team: Baoan Ji, M.D., Ph.D., Nicole R. Murray, Ph.D.
Central hypothesis: The KRAS gene encodes two proteins from alternative messenger RNA processing, and these two transcripts differ by two alternative fourth exons (4A and 4B) that encode the hypervariable region of these proteins. These proteins possess distinct posttranslational modifications, suggesting nonredundant functions. However, the evidence for their distinct activities in the literature is limited and often contradictory. This project aims to elucidate the molecular mechanisms of KRAS isoforms 4A and 4B during lung cancer initiation and progression.
Using CRISPR-Cas9-mediated genome editing, the investigative team recently created genetic mouse models that express either mutant KRAS4A or mutant KRAS4B. This technology allows the team to precisely target the KRAS4A or KRAS4B splicing acceptor sites, leading to the disruption of RNA splicing and protein expression. These novel genetic mouse models allow the research team to directly dissect the distinct roles of KRAS4A and KRAS4B during lung cancer development.
Potential outcomes or advances: Oncogenic KRAS gene mutations frequently occur in patients with lung cancer and are associated with a poor prognosis. Additionally, they often indicate a negative response to chemotherapy. So far, KRAS has been proved to be an intractable target.
Successful completion of the project's aims will provide new mechanistic insights into KRAS isoforms in lung cancer initiation and progression. Results will facilitate the development of anti-KRAS drugs for precision therapy by considering the distinct signaling of the two KRAS splice variants.
Title: Identifying novel molecular factors and pathways responsible for the leukemia-protective effects of the bone marrow microenvironment
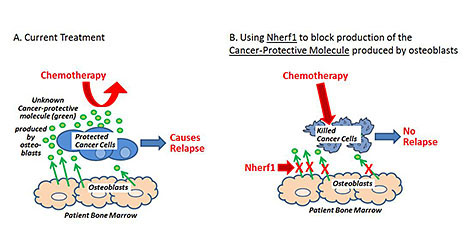
(A) Bone marrow osteoclasts produce chemoprotective molecules (green), which promote relapse. (B) Targeting the production of these molecules through Nherf1 could impact patient outcomes.
Investigative team: Karen E. Hedin, Ph.D., Scott H. Kaufmann, M.D., Ph.D., Jennifer Jane (Jennifer) Westendorf, Ph.D.
Central hypothesis: Relapse after cancer treatment is a heartbreaking problem for patients with leukemia, lymphoma, breast, prostate or other cancers. Relapse is often caused by cancer cells that survive chemotherapy because they metastasized into the bone marrow. Unfortunately, scientists do not understand how the bone marrow protects cancer cells from chemotherapy and therefore cannot prevent it from happening.
The investigative team has previously discovered that a type of normal cell in the bone marrow (osteoblasts) secrete an unknown cancer-protective molecule that — in the laboratory — protects leukemia cancer cells from chemotherapy. In addition, the team discovered that the Nherf1 protein blocks production of the cancer-protective molecule.
The team's results so far suggest that drugs that block the cancer-protective molecule or increase Nherf1 might be helpful to prevent relapse in patients being treated for cancer. The aim of this project is to achieve the following goals, which must be accomplished before testing such drugs in people:
- Determine the exact chemical structure of the cancer-protective molecule
- Determine why Nherf1 blocks production of the cancer-protective molecule
- Test if Nherf1 blocks the cancer-protective molecule in mice with leukemia, thus reducing relapse in the mice after chemotherapy
Potential outcomes and advances: This project is designed to define the chemical structure of the cancer-protective molecule and show how to block it in a living animal or person. These experiments are necessary before doctors can use this information to try to help prevent relapse in people being treated for cancer.
Title: Super-enhancer regulated senescent cell identity
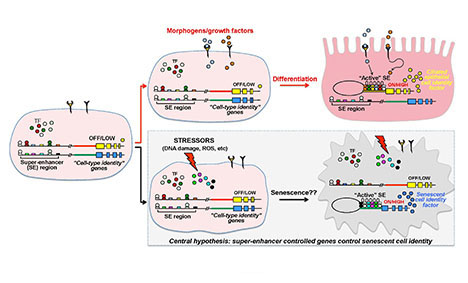
Senescent cells enter into a quiescent program leading to the transcription of genes that define the phenotype. These genes are likely coordinated by a "master" super-enhancer. Identification of the super-enhancer and associated transcription factors might reveal novel drug targets to eradicate senescent cells.
Investigative team: Hu Li, Ph.D.
Central hypothesis: This project aims to advance understanding of the gene regulatory mechanism that operates as a master switch to transform stressed cells into worn-out "zombie" (senescent) cells. This master switch is thought to work through the creation of super-enhancers, large stretches of DNA that recruit vast amounts of gene transcription regulators that switch on cell identity genes — genes that determine whether a cell becomes a brain, blood, liver, muscle or other specialized cell.
The investigative team's preliminary studies suggest that there is indeed a master switch for senescence. The specific goal of this project is to extend those studies by characterizing the genes that constitute the master switch and the cellular functions they control.
Senescent cells accumulate with age in many organs and tissues, releasing bioactive molecules that can harm neighboring cells. Senescent cells are also found at sites of pathology in several major human diseases, including cancer, cardiovascular disease, osteoarthritis and dementia.
Young adults virtually lack senescent cells, suggesting that they are not an essential cell type. This raises the possibility that the removal of senescent cells (senolysis) may be beneficial. In widely publicized studies, the investigative team demonstrated that periodic drug-induced elimination of senescent cells extends the healthy life span of laboratory mice. Read related publications on PubMed.
This indicates that treatments aimed at killing off these cells, or blocking their detrimental effects, might also help to combat aging and age-related diseases in humans. Currently, researchers are at the advent of clinical trials testing whether drug-induced removal of senescent cells benefits patients with osteoarthritis.
Potential outcomes or advances: By characterizing the genes that constitute the master switch and the cellular functions they control, the research team expects to unlock the inner workings of senescent cells at a molecular level, and with that their vulnerabilities.
Thus, in addition to uncovering a wealth of interesting and conceptually innovative biology highly relevant to aging and age-related diseases, the team expects to find important molecular entry points for the development of new classes of safe drugs that extend healthy life span by selectively killing detrimental senescent cells or by neutralizing the harmful molecules they secrete.
Title: Mechanisms of cardiac maladaptation in tumorigenic progeroid syndrome
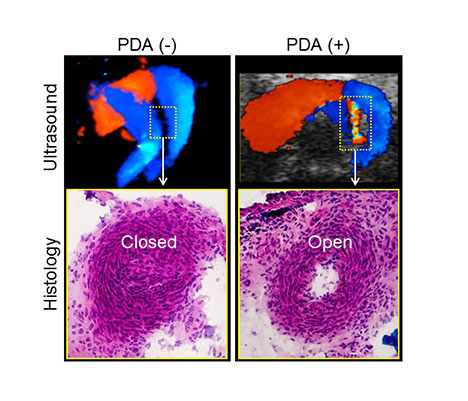
The disease pathway and a novel therapeutic target are investigated in a first-of-a-kind PDA model at functional (ultrasound; top panels), structural (histology; bottom panels) and molecular levels.
Investigative team: Satsuki Yamada, M.D., Ph.D., Sungjo Park, Ph.D., Darren J. Baker, M.S., Ph.D., Andre Terzic, M.D., Ph.D.
Central hypothesis: Ductus arteriosus is a blood vessel important in fetal development that naturally closes at birth. Failure of ductus closure, termed patent ductus arteriosus (PDA), compromises cardiopulmonary function after birth. Mechanisms of PDA are largely unknown, limiting therapeutic options.
Dr. Yamada and her investigative team at Mayo Clinic discovered a linkage between PDA and a regulator of aneuploidy, a state of abnormal chromosome numbers. Thus, they hypothesized that interventions that target aneuploidy effectors could rescue PDA. This central research question is comprehensively addressed by a multidisciplinary team.
Potential outcomes and advances: PDA affects 1 out of every 1,000 otherwise healthy newborns, or a quarter-million living Americans. This proof-of-concept study tests new therapies to rescue the incidence, severity and consequences of PDA, advancing the field toward a curative approach for PDA.
Title: Cooperative roles of loss of function of GAS7 and overexpression of MYCN in neuroblastoma dissemination and bone metastasis
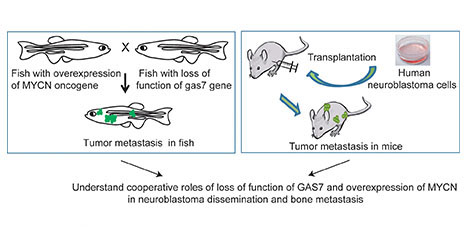
The goal of this project is to use both zebrafish and mouse models to understand the cooperative role of loss of function of GAS7 and overexpression of MYCN in neuroblastoma dissemination and bone metastasis.
Investigative team: Shizhen (Jane) Zhu, M.D., Ph.D., Saravut (John) J. Weroha, M.D., Ph.D., Jennifer Jane (Jennifer) Westendorf, Ph.D.
Central hypothesis: In this project, the investigative team is transplanting GAS7-suppressed human neuroblastoma cells into immunodeficient mice and examining bone metastasis using state-of-the-art imaging and histology analyses.
This research complements the team's previously developed zebrafish model of neuroblastoma metastasis. It is aimed at better understanding the multistep cellular and molecular pathogenesis, colonization and propagation of metastatic neuroblastoma cells in the bone. A second goal is to establish reliable in vivo zebrafish models of the aberrant genes and pathways that contribute to neuroblastoma metastasis.
Neuroblastoma is the most common extracranial solid tumor in children and accounts for approximately 12 percent of pediatric cancer deaths. About half of all patients, especially those older than 18 months, have evidence of metastasis at diagnosis. They experience a high risk of treatment failure and death, despite receiving greatly intensified chemotherapy.
The investigative team developed a novel zebrafish model of neuroblastoma metastasis by overexpressing the human MYCN oncogene and knocking out the GAS7 gene. Metastasis in this model occurs very early — within 4 months of age — and naturally, with widespread tumor cells disseminated to distant body parts, including the bone, which is the site of metastasis in approximately 60 percent of cases and causes extreme pain in patients.
The team's zebrafish model affords unique opportunities to study the molecular basis of neuroblastoma metastasis in vivo and to identify novel genes and pathways that cooperate with MYCN overexpression or GAS7 loss to promote this fatal stage of disease development.
Projected outcomes and advances: This project is expected to reveal how loss of function of GAS7 and overexpression of MYCN contribute to neuroblastoma dissemination and bone metastasis and to identify novel molecular targets that can be exploited therapeutically.
Additionally, the zebrafish models established in this project will be used in the near future to screen for effective small molecule inhibitors that block specific steps in metastasis with only minimal toxicity to normal tissues, and thus would be assigned high priority as candidate therapeutic agents.
Title: A novel mechanism generating resistance to EGFR inhibitors in lung cancer
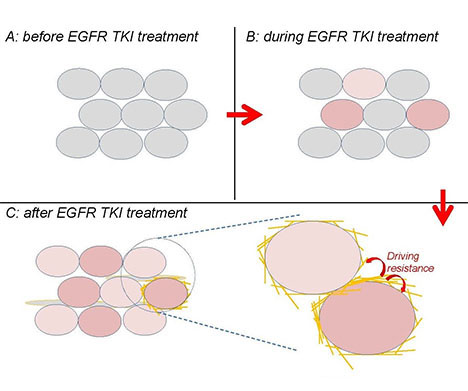
A novel mechanism generating resistance to EGFR tyrosine kinase inhibitors (TKIs) in lung cancer. A collagen-modifying enzyme is induced by EGFR TKI treatment, drives resistance through intracellular and intercellular signaling, and may serve as a novel therapeutic agent.
Investigative team: Ping Yang, M.D., Ph.D., Tobias Peikert, M.D.
Central hypothesis: Resistance to EGFR inhibitors remains a critical problem in lung cancer clinics: It inevitably and quickly develops in patients, and curative therapeutics treating resistance are lacking.
Building on the basis of the investigative team's discovery that EGFR inhibitors induce a collagen-modifying enzyme to drive resistance, this project investigates how this enzyme is induced by EGFR inhibitors and generates resistance, and whether its expression associates with clinical resistance and worse treatment outcomes.
Potential outcomes and advances: This project is expected to not only advance the understanding of the biology of resistance but also form the basis for future studies to test whether inhibiting the collagen-modifying enzyme is a useful strategy in treating resistance to EGFR inhibitors.